
Разделы статьи:
Ангиотензин II: ключевой продукт системы РААС
Ангиотензин II — это короткий пептид (всего 8 аминокислот), но по сути — гормон, один из самых мощных регуляторов артериального давления и объёма циркулирующей крови. Его действие комплексное, но имеет одну цель: быстро повысить и затем стабилизировать артериальное давление. Все. По факту, вся работа РААС по регуляции артериального давления упирается в производство и поддержание концентрации ангиотензина II. Давайте рассмотрим физиологические эффекты этого пептида.
Точки приложения ангиотензина II
Физиологическое действие ангиотензина II разностороннее. Однако все механизмы этого влияния приводят в конце концов к повышению АД. Что он конкретно делает?
- 1
Вазоконстрикция (самый быстрый эффект): сужение артериол, тем самым ангиотензин II увеличивает общее периферическое сосудистое сопротивление (ОПСС). Итог: быстрое повышение артериального давления. Это ключевой механизм, через который работает механизм РААС: повышение ангиотензина II → растет сосудистый тонус.
- 2
Влияние на почки: удержание натрия и воды. Он усиливает реабсорбцию натрия в проксимальных канальцах почек, вода следует за натрием, что приводит к увеличению объема циркулирующей крови (ОЦК). Повышенный ОЦК в сосудах = рост АД.
- 3
Активация симпатической нервной системы: усиливает выброс норадреналина из пресинаптических окончаний, снижает его обратный захват, действует центрально (ЦНС). Как результат: тахикардия (вспоминаете, как сердце начинает стучать в горле?), дополнительная вазоконстрикция → рост АД.
- 4
Стимулирует высвобождение альдостерона из коры надпочечников: ангиотензин II действует на zona glomerulosa надпочечников → стимулирует синтез альдостерона. Альдостерон – основной гормон, который увеличивает реабсорбцию натрия и воды в почках, приводя все к тому же увеличению ОЦК. Возникает разумный вопрос: зачем ему понадобился двойной механизм действия – через проксимальные канальцы почек + через альдостерон. Дело в том, что влияние на проксимальные канальцы – быстрое (минуты), а вот через альдостерон – это часы/дни поддержания АД…
- 5
Вазопрессин (он же – антидиуретический гормон АДГ): ангиотензин II стимулирует высвобождение вазопрессина: усиливается реабсорбция воды в собирательных трубочках, уменьшается диурез. Дополнительное удержание жидкости в конечном отделе нефрона → рост ОЦК.
- 6
Центры жажды (ЦНС): активирует гипоталамические центры, усиливает чувство жажды. Поведенческий эффект: человек начинает больше пить, что также увеличивает ОЦК.
Одним словом: все эффекты ангиотензина II на артериальное давление так или иначе связаны с прямым/опосредованным влиянием на вазоконстрикцию артериол или многофакторным увеличением ОЦК. Если уровень ангиотензина II будет постоянно повышенным – нет шансов, что организм не отреагирует повышением системного АД. Именно поэтому все «пляски с бубнами» в современной кардиологии и терапии крутятся вокруг этого гормона и РААС. Это не амлодипин или тиазидный диуретик с контролем АД по 1 механизму, тут разговор о 6 целях воздействия.
Как работает ангиотензин II
Действие РААС реализовано через ангиотензин II, активно повышающий давление крови через 6 разных механизмов. Но ведь влияние и на сосудистую стенку (вазоконстрикция), и на почки (удержание натрия и воды) и все прочие влияния осуществляется через какой-то клеточный рецептор. Ангиотензин II в данном случае выступает как тот фактор, который запускает сигналинг от него. В биохимии субстрат, который соединяется с конкретной мишенью на поверхности клетки, называют лигандом данного рецептора.
Если сказать совсем по-научному: ангиотензин II является эндогенным лигандом для рецепторов влияющих на АД и ОЦК. А их, как оказывается, ключевых — два: AT1 и AT2. Но прессорные эффекты преимущественно реализованы через AT1-рецепторы. Вот он наш главный рецептор-посредник – это АТ1 (Angiotensin II Type 1 receptor = рецептор ангиотензина II, тип 1).
При его активации, а это классический G-белковый рецептор, он изменяет функцию белков через их фофорилирование протеинкиназой С (Protein Kinase C). Это – научная составляющая, а если по простому – то при стимуляции АТ1 рецепторов клетки разных тканей начинают работать иначе — или сокращаются гладкомышечные волокна, или активируют захват натрия, или стимулируют определенные элементы других систем. Т.е. конечная схема: ангиотензин II → АТ1 рецептор → Protein Kinase C → изменение функциональной активности клетки → повышение системного АД.
Но не надо думать, что ангиотензин II – это сплошное зло. Пока его концентрация колеблется в физиологическом коридоре, он – тот прецизионный, тонкий, чуткий помощник, который держит давление, обеспечивает нормальную скорость клубочковой фильтрации, поддерживает гомеостаз и кислотно-щелочное равновесие. РААС – это уникальная и точная система поддержания жизнедеятельности.
Но совершенно другое дело, когда РААС находится в состоянии постоянной гиперактивации, и уровень ангиотензина II превышает физиологические флуктуации в течение длительного времени. Проблема возникает, когда система, рассчитанная на минуты–часы, работает месяцами–годами. Именно тогда создаются условия для ремоделирования сосудов (теряется эластичность), гипертрофии миокарда, прогрессирования фиброза. РААС — это высокоточная система выживания, которая при хронической активации превращается в механизм повреждения органов-мишеней.
Вот тут самое время сказать про весьма интересный рецептор АТ2, упомянутый мною ранее. Ангиотензин II с одинаковой силой связывается с обоими рецепторами: и АТ1, и АТ2. На языке науки – аффинитет к рецепторам у него одинаковый. Но действие АТ2 рецептора на клеточном уровне – противостоять влиянию AT1. Стимуляция АТ2 приводит к расширению сосудов, замедлению ремоделирования и другим эффектам прямо противоположным влиянию АТ1 сигналинга. Разумный вопрос: а почему тогда ангиотензин II вообще работает и оказывает эффект? Ведь АТ2 может просто уравновесить его эффект. Нет, не может, и вот почему.
Все дело в концентрации рецепторов на единицу площади (так называемой экспрессии), типа сигналинга и физиологической роли. У взрослых людей экспрессия АТ2 рецептора ограничена — массовый ответ формирует AT1 просто за счёт численного доминирования. Кроме этого, АТ2 сигналинг представлен не классическим быстрым G-белковым путем, а нетипичными путями активации клеток, он просто более медленный. Ну и наконец АТ2 –рецептор – это своего рода регулятор, тормоз: ограничить рост, снизить тонус, противодействовать ремоделированию. Поэтому AT2 физиологически не должен доминировать — иначе система не выполнит свою основную задачу. Иными словами, назначение АТ2 – сгладить пики влияния ангиотензина II, противостоять его повышению до определенной степени.
Почему я об этом говорю? Это будет достаточно важно в контексте применения сартанов – они заблокируют АТ1 рецептор, оставив свободным АТ2 и совершенно не тронув ангиотензин II. Таким образом, они не только полностью снимут прессорное влияние через АТ1 рецептор, но и будут стимулировать восстановление органов-мишеней через стимуляцию АТ2. Это важно при подборе препаратов в конкретных клинических контекстах!
Теперь мы хорошо понимаем, что РААС работает через ангиотензин II и стимуляцию АТ1 рецептора, реализующих 6 разных механизмов повышения АД. Мы так же понимаем, что есть АТ2 рецептор, который выступает как небольшой регулятор отрицательных действий ангиотензина II, возникающих при хронизации состояния. Он эффективно противостоит пагубному влиянию, если физиологические концентрации ангиотензина II превышают норму временно, но ничего не может сделать, если это превышение – постоянное.
РААС в подробностях
Сейчас самое время разобраться, как работает сама система и как ангиотензин II синтезируется. Заодно и посмотрим на наших «героев» — прилы/сартаны, поймем суть их работы. Ничего хитрого, но…
В фокусе нашего внимания будут почки. Почка в контексте РААС выступает главным сенсорным органом, ангиотензин II — центральным механизмом влияния, а сосуды, сердце и те же почки — мишенями. Начну конечно с анатомии и гистологического среза клубочкового аппарата почек – там все начинается.
Кровь поступает в нефрон через приносящую (афферентную) артериолу, которая входит в капсулу клубочка и распадается на сеть капилляров — гломерулярный клубочек. Здесь происходит фильтрация плазмы с образованием первичной мочи. Затем кровь покидает клубочек через выносящую (эфферентную) артериолу. Важная особенность: диаметр и тонус приносящей и выносящей артериол регулируются независимо, что позволяет тонко настраивать внутриклубочковое давление и скорость фильтрации. Приносящая артериола сильно реагирует на аденозин через А1-рецепторы: это вызывает ее сужение и падение давления на входе в клубочек, перфузия клубочка уменьшается, падает скорость клубочковой фильтрации (СКФ).
Выносящая артериола на аденозин практически не реагирует, поэтому аденозин – специфический регулятор поступления крови в клубочек. Но выносящая артериола имеет гораздо больше АТ1 рецепторов, чем приносящая – при равной концентрации ангиотензина II выход из клубочка будет заблокирован больше чем вход, давление в клубочке вырастет, фильтрация (СКФ) повысится. Ангиотензина II – специфический регулятор оттока крови из клубочка.
После фильтрации первичная моча проходит по выводящим канальцам нефрона. Вот что интересно – канальцы, по которым выводится первичная моча, делают изгиб и на обратном пути проходят рядом с клубочком. В области, где расположен дистальный извитой каналец, он проходит вплотную между приносящей и выносящей артериолами своего же клубочка. Получается, что этот участок канальца отводящего мочу прилежит к основанию фильтрующего клубочка. И это не просто так: в этом месте располагается специализированная зона — MACULA DENSA. Это компактный участок эпителиальных клеток дистального канальца, ориентированный на «сканирование» состава мочи, прежде всего концентрацию NaCl.
А теперь внимание: приносящая артериола, как манжетой, обернута специализированными юкстагломерулярными (ЮГ) клетками — это модифицированные гладкомышечные клетки, способные секретировать ренин. Выносящая артериола лишена этих клеток, но прямо к месту между приносящей и выносящей артериолой примыкает обратная сторона чувствительной Макулы Денса, которая своими рецепторами (лицом) смотрит в мочу канальцев. Между ЮГ-клетками и макулой денса расположены экстрагломерулярные мезангиальные клетки, обеспечивающие передачу сигналов внутри комплекса. Таким образом, анатомически формируется «треугольник»: приносящая артериола (с ЮГ-клетками) — выносящая артериола — Макула Денса дистального канальца и между ними передатчики сигналов – мезангиальные клетки. Поздравляю – я только что описал всю структуру сложного юкстагломерулярного почечного комплекса («юкста» – означает рядом, т.е. рядом с клубочком). Но как это все работает?
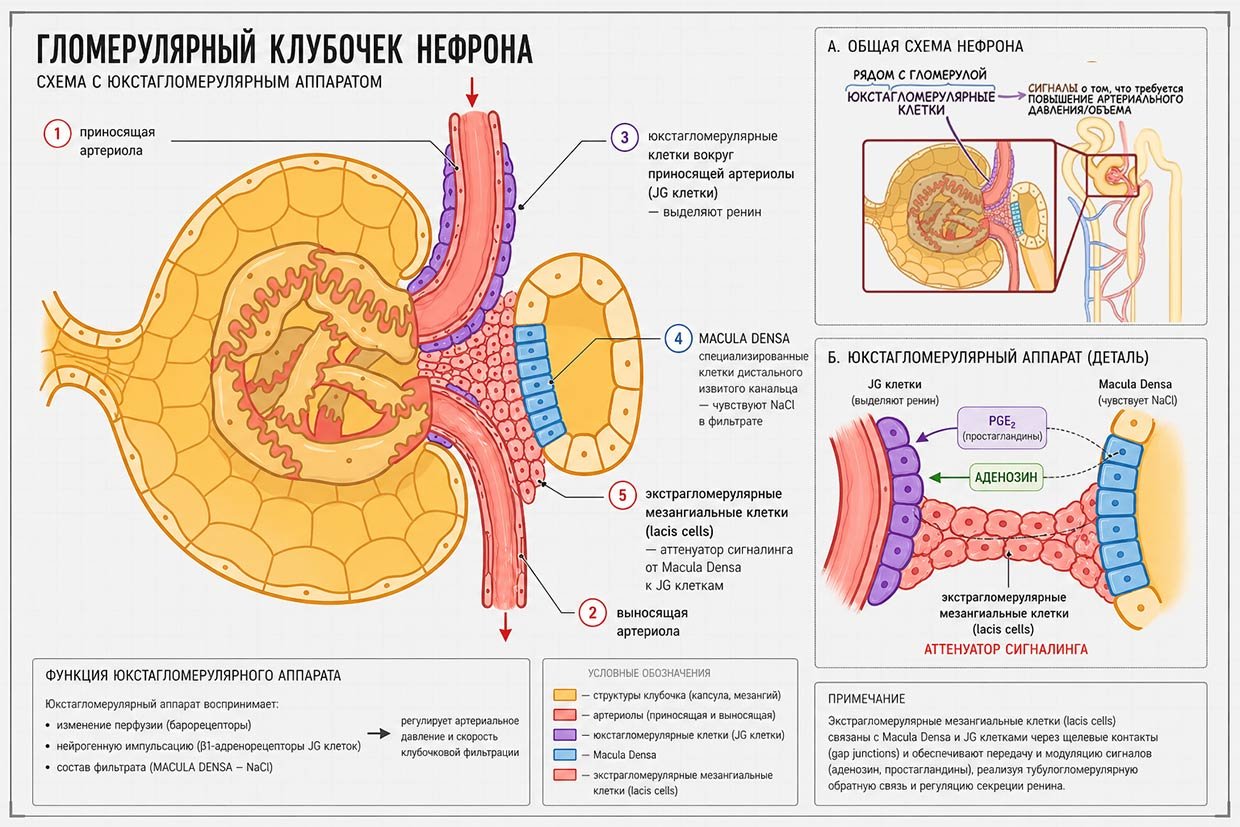
Юкстагломерулярные клетки как центральный сенсор и продуцент ренина
ЮГ-клетки уникальны тем, что объединяют сенсорную и секреторную функции. Они реагируют на три типа стимулов.
Гемодинамический путь. Если давление крови падает, то снижение давления в приносящей артериоле уменьшает растяжение её стенки. ЮГ-клетки, окутывающие манжетой эту артериолу, через свои барорецепторы воспринимают это как сигнал дефицита перфузии и увеличивают секрецию ренина, гранулы которого они и содержат. Это локальный «внутрипочечный барорецептор», работающий независимо от системных датчиков.
Нейрогенный путь. Системные барорецепторы от дуги аорты и каротидного синуса при падении давления увеличивают симпатическую импульсацию. И… эти нервные импульсы имеют прямую связь с ЮГ-клетками! ЮГ-клетки располагают собственными β1-адренорецепторами, которые «чувствуют» падение системного давления, и это прямо стимулирует их к выбросу ренина. Таким образом, почка получает централизованный сигнал о снижении давления ещё до выраженных локальных изменений.
Канальцевый путь. Макула Денса оценивает концентрацию NaCl в просвете дистального канальца. Дело в том, что концентрация NaCl напрямую зависит от давления в клубочке и скорости клубочковой фильтрации (СКФ) – если СКФ большая (а это значит давление в клубочке большое), то и концентрация ионов натрия и хлора в моче будет большой, если СКФ маленькая (падение давления), то и концентрации натрия/хлора упадут.
При снижении концентраций Макула Денса выделяет простагландины (прежде всего PGE₂). Простагландины сигнализируют ЮГ-клеткам – а ну-ка поддайте ренина! При повышении NaCl, напротив, клетки Макулы Денса выделяют АТФ в интерстиций с мезангиальными клетками, который превращается в аденозин (простагландины блокируются). Через аденозин реализуется тормозящий сигнал и на ренин, и на фильтрацию, а именно: приносящая артериола сужается, давление в приносящей системе падает, это вызывает подавление фильтрации и концентрация NaCl падает.
Таким образом, в одной точке сходятся три принципиально разных сигнала: давление, нервная регуляция и состав фильтрата. Это и делает почку ключевым «интегратором» гемодинамики. Важно, что эти три канала не конкурируют, а складываются: при истинной гиповолемии или гипотензии они активируются синхронно и потенцируют друг друга. Ключевой продукт «на выходе» — РЕНИН из ЮГ-клеток. А дальше – все предельно просто:
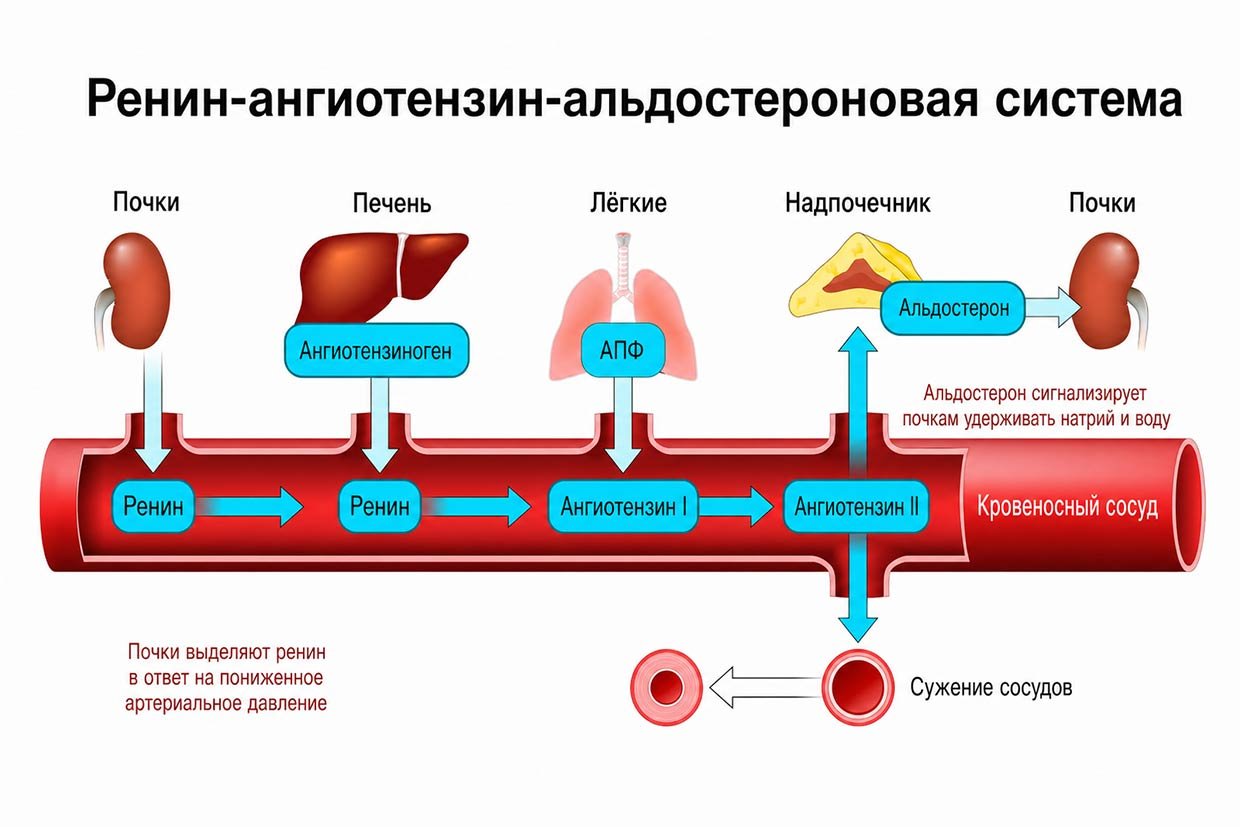
Ренин и ангиотензиноген: запуск протеолитического каскада
Попав в системный кровоток, ренин не действует «на систему в целом», а ведёт себя строго как специфический фермент с одной мишенью — найти ангиотензиноген. Ангиотензиноген – белок, постоянно синтезируется печенью и циркулирует в плазме в относительно стабильной концентрации, формируя своего рода «резерв субстрата» для быстрого запуска РААС.
Ренин распознаёт ангиотензиноген и отщепляет от огромной молекулы малюсенький, но определённый фрагмент — декапептид, состоящий из 10 аминокислот. Так образуется ангиотензин I – это пептид из 10 аминокислот.
Важно, что эта реакция является скорость-лимитирующим этапом всей системы: именно уровень и активность ренина в наибольшей степени определяют интенсивность дальнейшего каскада. Сам ангиотензиноген при этом редко является лимитирующим фактором, за исключением отдельных состояний (например, повышение его уровня под влиянием эстрогенов или глюкокортикоидов).
Ангиотензин I — это биологически относительно инертный пептид. Его основная роль — служить промежуточным субстратом для следующего этапа. Дальнейшая судьба ангиотензина I определяется действием ангиотензин-превращающего фермента (Angiotensin-converting enzyme=ACE). Это фермент (произносим: дипептидилкарбоксипептидаза :-) ), локализованный преимущественно на поверхности эндотелиальных клеток.
Классически подчёркивается роль лёгких (именно в них, как указывают многие источники, происходит дальнейшее превращение ангиотензина I), поскольку там огромная поверхность капиллярного русла обеспечивает максимально эффективный контакт крови с эндотелием. Однако принципиально важно, что фермент АПФ экспрессируется и в других сосудистых бассейнах — поэтому превращение ангиотензина I происходит по всему сосудистому руслу, а не только в лёгких.
АПФ отщепляет от ангиотензина I еще две аминокислоты, превращая его в октапептид из 8 аминокислот — ангиотензин II, который мы уже хорошо знаем. Вот и завершился наш каскад:
упало системное АД → отреагировала система РААС через ЮГ-клетки → Ренин → Ангиотензин I → Ангиотензин II → АД поднялось.
И надо сказать, что этот каскад очень эффективный: даже небольшое увеличение активности ренина быстро приводит к значимому росту концентрации ангиотензина II, поскольку последующие этапы протекают относительно быстро и не являются лимитирующими. Хорошо, а как же работают иАПФ препараты и сартаны?
Блокада РААС: две точки входа в один каскад
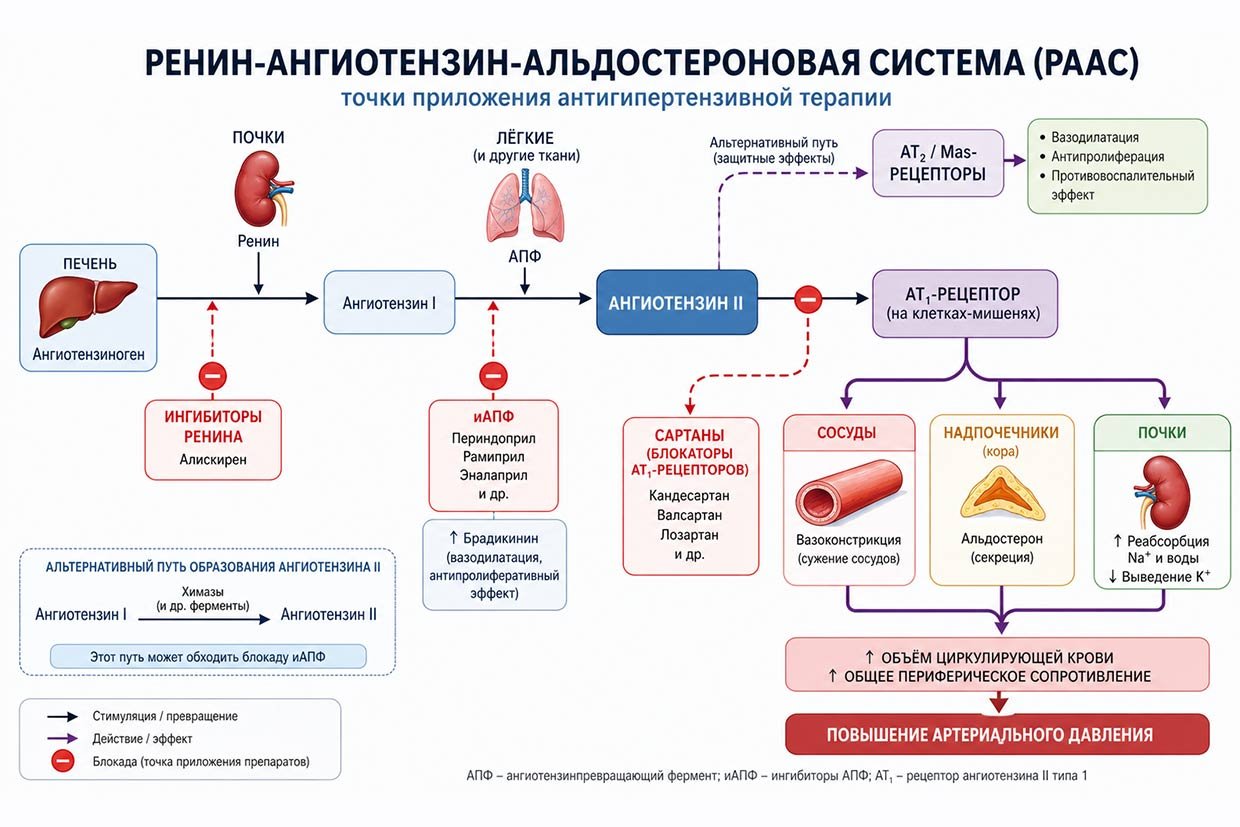
Ингибиторы ангиотензин-превращающего фермента (иАПФ/прилы) и блокаторы рецепторов ангиотензина II (сартаны, БРА) вмешиваются в один и тот же регуляторный контур, но на разных уровнях. В обоих случаях конечная цель — ослабить ангиотензин-зависимые эффекты, однако путь к ней принципиально различается.
Ингибиторы АПФ: блокада образования эффектора
Препараты класса иАПФ действуют на уровне ангиотензин-превращающего фермента (Angiotensin — converting enzyme), который катализирует превращение ангиотензина I в ангиотензин II.
иАПФ просто блокируют (выключают) этот фермент, что снижает синтез ангиотензина II. В результате падения концентрации ангиотензина II ослабевают его сосудистые и гормональные эффекты.
Все просто: блокируем АПФ, ангиотензин II просто не образуется из ангиотензина I. РААС активирована, но нет действующего начала — ангиотензина II.
Принципиально важно, что наш фермент АПФ участвует не только в образовании ангиотензина II, но и в деградации брадикинина (!). А брадикинин – специфический вазодилятатор. Поэтому ингибирование фермента приводит к накоплению брадикинина, что усиливает вазодилатацию за счёт высвобождения NO и простагландинов. Этот компонент делает эффект иАПФ более «широким», но одновременно объясняет специфические побочные реакции: накопление брадикинина в бронхах приводит к кашлю, а вокруг сосудов может вызвать редкое осложнение — ангиоотёк.
Сартаны: блокада рецепторного ответа
Тут все по-другому. Ангиотензин II образуется, как ему и положено. Далее он должен соединиться со своей мишенью – рецептором АТ1. Но сартаны (БРА) – это специализированные блокаторы этих АТ1 рецепторов (Angiotensin II receptor type 1 blockers). Т.е. их действие реализуется на уровне мишени, а не синтеза. Селективно блокируя сам рецептор, они тормозят все эффекты ангиотензина II.
Таким образом, даже при сохранённой или повышенной концентрации ангиотензина II:
- его взаимодействие с AT1-рецепторами блокировано
- сосудистые и гормональные эффекты не реализуются
При этом важно — ангиотензин II продолжает взаимодействовать с AT2-рецепторами, которые функционально противоположны AT1 (вазодилатация, антипролиферация). Это создаёт дополнительный «смещающий» эффект в сторону вазопротекции, ремоделирования в положительную сторону миокарда.
Главное различие между классами — уровень вмешательства в каскад РААС.
- иАПФ уменьшают образование ангиотензина II
- Сартаны блокируют реализацию его действия на рецепторе
И тут начинается самое интересное. Кроме общей системы РААС (почечной) существую локальные РААС. Да, да, я не ошибся – в сердце, почках, мозге и сосудистой стенке(!) есть собственные мини- РААС, которые занимаются все тем же – продуцируют ангиотензин II. Эта тканевая РААС функционирует полуавтономно:
- ангиотензин II может синтезироваться прямо в интерстиции;
- концентрации в тканях могут значительно превышать плазменные;
- эффекты реализуются «на месте», без обязательного выхода в системный кровоток.
Ключевой момент: в тканях путь образования ангиотензина II не ограничивается АПФ превращением из ангиотензина I. Это как? Помимо АПФ, превращение ангиотензина I в ангиотензин II может происходить с участием других ферментов, прежде всего химазы (сериновой протеазы, высвобождаемой тучными клетками). Этот путь активируется при повреждении, воспалении. Более того, в патологических условиях вклад химазы может становиться существенным или даже доминирующим, что означает одно — в тканях может продолжаться синтез ангиотензина II.
Что это меняет для иАПФ в клиническом контексте? Одно — системная активность РААС будет подавлена, но локальные эффекты (например, нехорошее ремоделирование миокарда или сосудистой стенки) могут сохраняться.
Сартаны выглядят иначе: неважно, как образовался ангиотензин II (через АПФ или химазу), если он достиг рецептора — его действие будет заблокировано. Поэтому сартаны обеспечивают более полную блокаду эффекторного звена. Более того: блокируя АТ1 рецептор, уровень ангиотензина II может даже повышаться. Посудите сами – РААС активирована, но не видит результата, она еще больше повышает уровень ангиотензина II за счет обратной связи. И… он начинает активнее взаимодействовать с AT2-рецепторами, а это:
- вазодилатация;
- антипролиферация;
- антифиброз;
Означает ли это, что сартаны предпочтительнее? И вот тут мы вдадимся уже в философские дискуссии. С одной стороны сартаны более полноценно заблокируют РААС, что конечно имеет значение. И отлично скажутся на долговременной защите от фиброзных изменений. С другой – блокада РААС до определенного уровня приносит результаты, выше – очень незначительные.
Но не надо забывать, что у иАПФ свои преимущества, которых совершенно нет у сартанов: повышение концентрации брадикинина увеличивает накопление NO и простагландинов в стенке сосуда = эндотелиально-зависимая вазопротекция, которая очень важна при атеросклерозе, тромбообразовании, воспалении. Ведь не одной АТ1 блокадой мир строится, погибает человек в конце концов от тромбов и дисфункции эндотелия, а иАПФ от этого защищают. Ну и еще каплю меда/дегтя – не знаю: у каждого человека распределение рецепторов разное, активность АПФ тоже, препараты по разному проникают в ткани. Все это во многом выравнивает действие этих смежных групп.
Ну и напоследок: у сартанов есть тоже эндотелиальная протекция, но она выражена слабее, чем брадикининовая у иАПФ. И я говорю о следующем механизме:
при работе сартанов концентрация ангиотензина II растет. Им надо куда-то деваться. Да они начинают максимально садиться на АТ2 с хорошими эффектами для организма, но … есть и другой путь утилизации избыточного ангиотензина II. У нас присутствует специализированный фермент под названием АСЕ-2: ангиотензин-превращающий-фермент-2 типа. Он начинает сбрасывать ангиотензин II в новую сущность: ангиотензин-(1–7). Вот именно так он и называется. Это – лиганд для рецептора Mas. Mas-рецептор обеспечивает:
- Вазодилатацию;
- ↑ NO;
- антифибротическое действие;
- противовоспалительный эффект;
- улучшение эндотелиальной функции.
Итоговые размышления
Существование тканевой РААС и альтернативных путей образования ангиотензина II, в частности с участием химазы, ограничивает эффективность ингибиторов АПФ на уровне синтеза гормона. В отличие от них, блокаторы AT1-рецепторов (БРА) обеспечивают более полную функциональную блокаду системы, независимо от источника ангиотензина II, одновременно сохраняя активацию вазопротективных AT2-рецепторов. Однако ингибиторы АПФ компенсируют эту особенность за счёт брадикининового компонента, быстро и мощно усиливающего эндотелиальную вазодилатацию, в отличие от медленного пути сартанов через Mas-рецепцию, которая тем не менее присутствует.


